Introduction
sanger-tol/curationpretext is a bioinformatics pipeline typically used in conjunction with TreeVal to generate pretext maps (and optionally telomeric, gap, coverage, and repeat density plots which can be ingested into pretext) for the manual curation of high quality genomes.
This is intended as a supplementary pipeline for the treeval project. This pipeline can be simply used to generate pretext maps, information on how to run this pipeline can be found in the usage documentation.
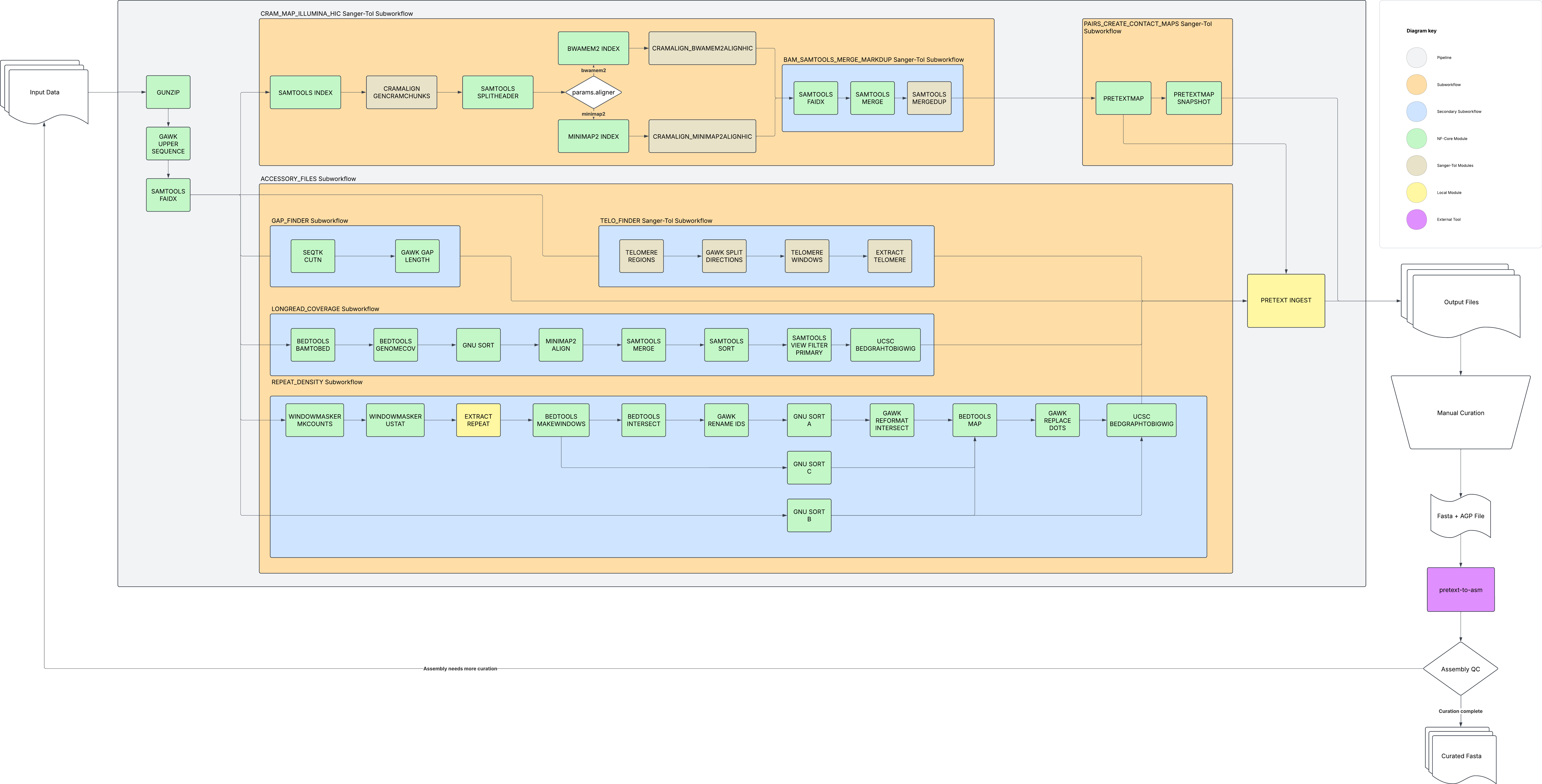
The above image shows the use of this pipeline inside of the manual curation process and follows the below major steps.
-
CRAM_MAP_ILLUMINA_HIC (ALIGN_CRAM) + PAIRS_CREATE_CONTACT_MAPS (CREATE_MAPS) - Generates pretext maps as well as a static image.
-
ACCESSORY_FILES - Generates the repeat density, gap, telomere, and coverage tracks.
-
PRETEXT_INGEST - Imports the generated tracks into pretext for visualisation.
Usage
[!NOTE]
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
[!NOTE]
If you are new to Nextflow and nf-core, please refer to this page on how to set-up Nextflow. Make sure to test your setup with -profile test before running the workflow on actual data.
Currently, the pipeline uses the following flags:
-
--input- The absolute path to the assembled genome in, e.g.,
/path/to/assembly.fa
- The absolute path to the assembled genome in, e.g.,
-
--sample- Sample is the naming prefix of the output files, e.g. iyTipFemo
-
--reads- The directory of the fasta files generated from longread reads, e.g.,
/path/to/fasta/ - This folder must contain files in a
.fasta.gzformat, or they will be skipped by the internal file search function.
- The directory of the fasta files generated from longread reads, e.g.,
-
--read_type- The type of longread data you are utilising, e.g., ont, illumina, hifi.
-
--aligner- The aligner you wish to use for the coverage generation, defaults to
AUTObut options includebwamem2andminimap2.
- The aligner you wish to use for the coverage generation, defaults to
-
--cram- The directory of the cram and cram.crai files, e.g.,
/path/to/cram/
- The directory of the cram and cram.crai files, e.g.,
-
--map_order- hic map scaffold order, input either
lengthorunsorted
- hic map scaffold order, input either
-
--teloseq- A telomeric sequence, e.g.,
TTAGGG
- A telomeric sequence, e.g.,
-
--multi_mapping- Level of multi-mapping read filtering to perform whilst building the pretext map.
-
--all_output- An option to output all maps + accessory files, the default will only output the pretextmaps where ingestion has occured.
-
--skip_tracks- A csv list of accessory tracks to skip, options are:
ALL,gap,coverage,telo,repeats,NONE. Default isNONE. Please note that capitalization matters.
- A csv list of accessory tracks to skip, options are:
-
--split_telomere- A boolean to also generate the telomere track in 5Prime and 3Prime styles, this is also include the original telomere track.
-
--pre_mapped_bam- A boolean option to use
--cramas input for A pre-mapped bam file.
- A boolean option to use
-
--cram_chunk_size- The number of records in a cram file should be chunked into, defaults to 10000.
-
--run_hires- A boolean to run the pipeline in hires mode, i.e., generate hires resolution maps. Default is
true
- A boolean to run the pipeline in hires mode, i.e., generate hires resolution maps. Default is
-
--run_ultra- A string argument to run the pipeline in ultra resolution mode, i.e., generate ultra resolution maps. Options are:
yes,force,no. Default isyes, this runs ultra resolution maps is the genome file is > 4.Gb.
- A string argument to run the pipeline in ultra resolution mode, i.e., generate ultra resolution maps. Options are:
-
--snapshot_order- A path to a
genome,sizesorfaifile containing the scaffolds in the order required for the output snapshot png file.
- A path to a
Now, you can run the pipeline using:
nextflow run sanger-tol/curationpretext \
--input { input.fasta } \
--cram { path/to/cram/ } \
--reads { path/to/longread/fasta/ } \
--read_type { default is "hifi" }
--sample { default is "pretext_rerun" } \
--teloseq { default is "TTAGGG" } \
--map_order { default is "unsorted" } \
--multi_mapping { default is "0" (for no filtering of multi-mapping reads)} \
--all_output <true/false> \
--outdir { OUTDIR } \
-profile <docker/singularity/{institute}>
Warning: Please provide pipeline parameters via the CLI or Nextflow
-params-fileoption. Custom config files including those provided by the-cNextflow option can be used to provide any configuration except for parameters;
For more details, please refer to the usage documentation and the parameter documentation.
Pipeline output
To see the the results of a test run with a full size dataset refer to the results tab on the sanger-tol/curationpretext website pipeline page. For more details about the output files and reports, please refer to the output documentation.
Credits
sanger-tol/curationpretext was originally written by Damon-Lee B Pointon (@DLBPointon).
We thank the following people for their extensive assistance in the development of this pipeline:
-
@muffato - For reviews.
-
@yumisims - TreeVal and Software.
-
@weaglesBio - TreeVal and Software.
-
@josieparis - Help with better docs and testing.
-
@mahesh-panchal - Large support with 1.2.0 in making the pipeline more robust with other HPC environments.
-
@GRIT - For feedback and feature requests.
-
@prototaxites - Support with 1.3.0 and showing me the power of GAWK.
Contributions and Support
If you would like to contribute to this pipeline, please see the contributing guidelines.
Citations
If you use sanger-tol/curationpretext for your analysis, please cite it using the following doi: 10.5281/zenodo.12773958
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
This pipeline uses code and infrastructure developed and maintained by the nf-core community, reused here under the MIT license.
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.







