Introduction
This document describes the output produced by the TreeVal pipeline.
The directories listed below will be created in the results directory after the pipeline has finished. All paths are relative to the top-level results directory.
Pipeline overview
The pipeline is built using Nextflow and processes data using the following workflows:
-
generate-genome - Builds a genome description file of the reference genome.
-
read-coverage - Produces read coverage based on hifi/clr/ont/illumina fasta file/s.
-
gap-finder - Identifies gaps in the input genome.
-
repeat-density - Reports the intensity of regional repeats within an input assembly.
-
hic-mapping - Aligns illumina HiC short reads to the input genome, generates mapping file in three format for visualisation:
.pretext,.hicand.mcool. -
telo-finder - Identifies regions of a user given telomeric sequence.
-
gene-alignment - Aligns the peptide and nuclear data from assemblies of related species to the input genome.
-
insilico-digest - Generates a map of enzymatic digests using 3 Bionano enzymes.
-
selfcomp - Identifies regions of self-complementary sequence.
-
synteny - Generates syntenic alignments between the input and other high quality genomes.
-
busco-analysis - Uses BUSCO to identify ancestral elements. Also use to identify ancestral Lepidopteran genes (merian units).
-
kmer - Counts k-mer and generates a copy number spectra plot.
-
pretext-ingestion - Ingests accessory files into the pretext file.
-
pipeline-information - Report metrics generated during the workflow execution
Note
Some terms used here are fairly Sanger specific, for example punchlists.
Punchlists are bed files containing sites of interest:
gap_punchlist.bedcontains the coordinates of gap regions in the genome.{species}_cdna_punchlist.bedcontains the coordinates of a cDNA aligments, cDNA being sourced from the{species}.
This same logic can be applied to all punchlists. We will work on making these more generic, however, as they are used in downstream processes that require seperate updates they will remain for now.
generate-genome
This workflow generates a .genome file which describes the base pair length of each scaffold in the reference genome. This file is then recycled into the workflow to be used by a number of other subworkflows.
Output files
hic_files/{sample}.sizes: Chromosome ID and length description file of the input genome.

read-coverage
Read Coverage uses genome sequence reads (HiFi, CLR, ONT or Illumina) reads to generate a coverage bigWig as well as a trio of depth.bigbed files.
Output files
-
hic_files/{sample}_coverage_normal.bigWig: Coverage of aligned reads across the reference genome in bigwig format.
-
treeval_upload/punchlists/maxdepth.bed: Max read depth punchlist in bed format (max refers to regions where read coverage exceeds 1000).zerodepth.bed: Zero read depth punchlist in bed format.halfcoverage.bed: Half read depth punchlist in bed format.
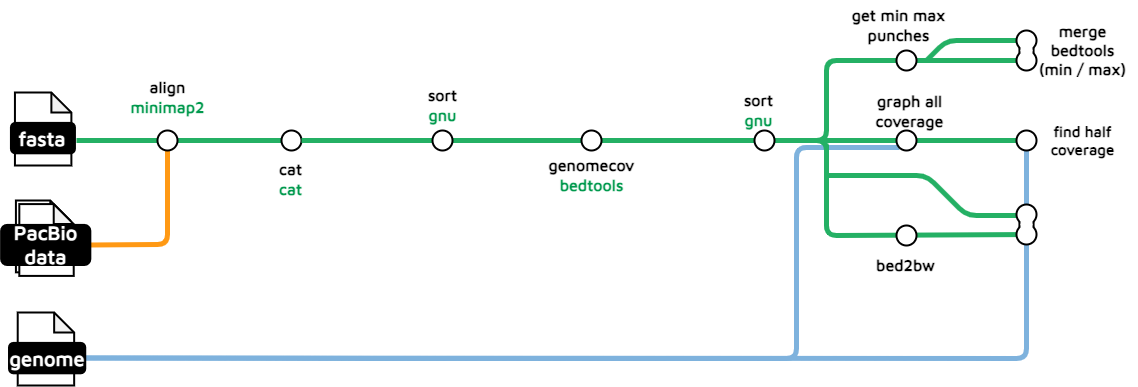
gap-finder
The gap-finder subworkflow generates a bed file containing the genomic locations of the gaps in the sequence. This file is injected into the hic_maps at a later stage. The output bed file is then BGzipped and indexed for display on JBrowse.
Output files
treeval_upload/gap_{sample}.bed.gz: A bgzipped file containing gap locations*.bed.gz.csi: An index file for the above file in Coordinate-Sorted Index format.
hic_files/{sample}_gap.bed: The raw bed file needed for ingestion into Pretext

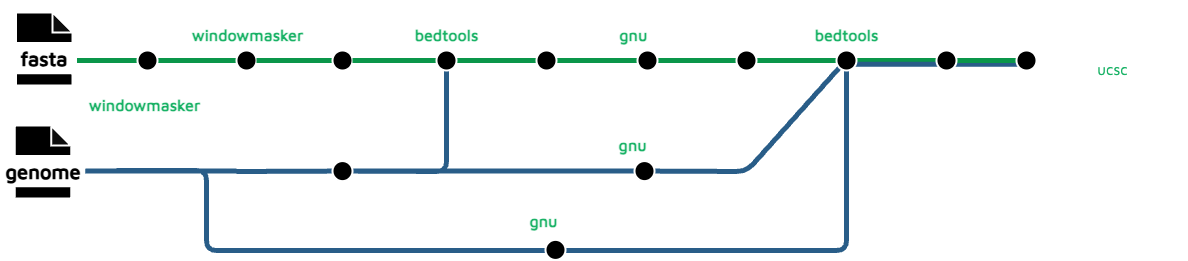
repeat-density
This uses WindowMasker to mark a repeat distribution across the input genome. The genome is chunked into 10kb bins which move along the entire genome as sliding windows in order to profile the repeat intensity. These fragments are then mapped back to the original assembly for visualisation purposes.
Output files
hic_files/{sample}_repeat_density.bigWig: Intersected read windows aligned to the reference genome in bigwig format.
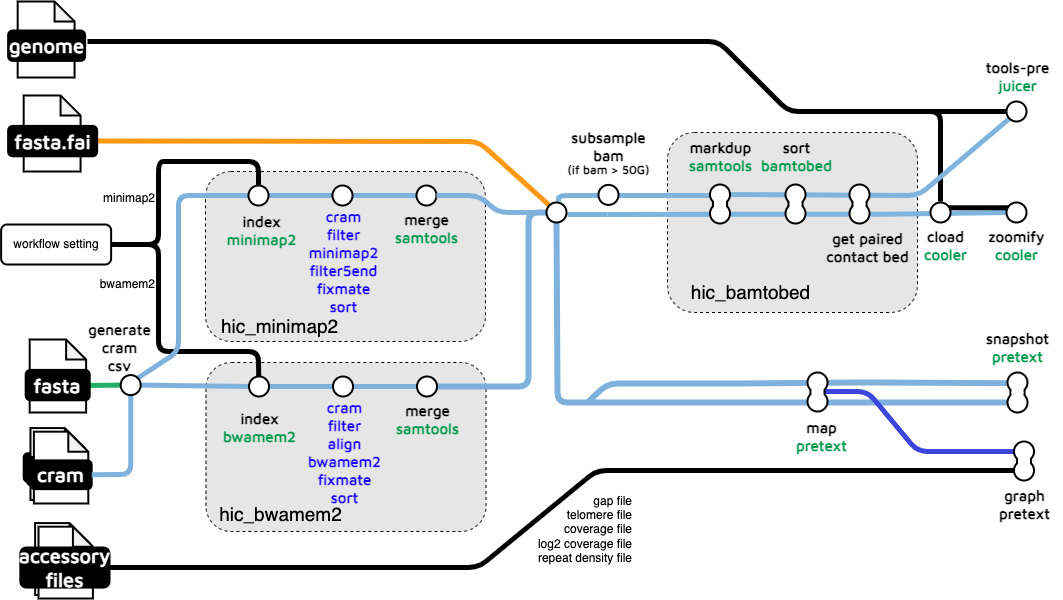
hic-mapping
The hic-mapping subworkflow takes a set of HiC read files in .cram format as input and derives HiC mapping outputs in .pretext, .hic, and .mcool formats. These outputs are used for visualization on PretextView, Juicebox, and HiGlass respectively.
Output files
hic_files/*_pretext_ultra.pretext: Ultra resolution pretext map.*_pretext_hr.pretext: High resolution pretext map.*_pretext_normal.pretext: Standard resolution pretext map.*.mcool: HiC map required for HiGlass*_normalFullMap.png: A pretext snapshot of the normal resolution pretext ONLY. Making snapshots of the other pretexts becomes very computationally expensive for no real gain in quality.
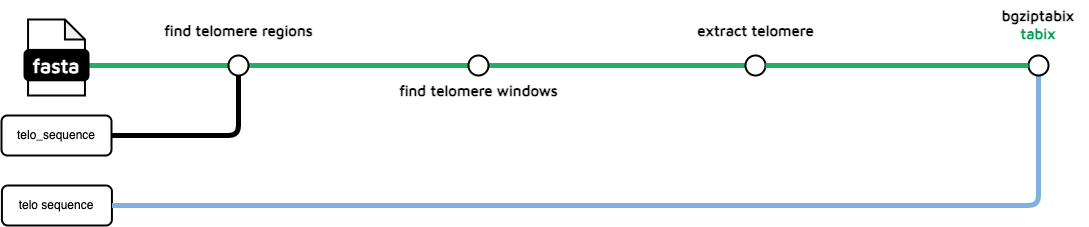
telo-finder
The telo-finder subworkflow uses a supplied (by the .yaml) telomeric sequence to identify putative telomeric regions in the input genome. The BGZipped and indexed file is used in JBrowse and as supplementary data for HiGlass and PreText.
Optionally, you can now also use the --split_telomere flag to generate split telomere files. These are telomeres in the 3 and 5 prime directions. These files will have 3P or 5P in their name.
Output files
treeval_upload/telo_*.bed.gz: A bgzipped file containing telomere sequence locationstelo_*.bed.gz.csi: A csi index file for the above file.
hic_files/*_telomere.bed: The raw .bed file needed for ingestion into Pretext*_5P_telomere.bed: Containing the forward strand associated telomere sites.*_3P_telomere.bed: Containing the reverse strand associated telomere sites.
busco-analysis
The BUSCO annotation subworkflow takes an assembly genome as input and extracts a list of BUSCO genes based on the BUSCO results obtained from BUSCO. Additionally, it provides an overlap BUSCO gene set based on a list of lepidoptera ancestral genes (Wright et al., 2023), which has been investigated by Charlotte Wright from Mark Blaxter's lab at the Sanger Institute.
Output files
treeval_upload/*_buscogene.bigbed: BigBed file for BUSCO genes track.*_ancestral.bigbed: BigBed file for ancestral elements track.
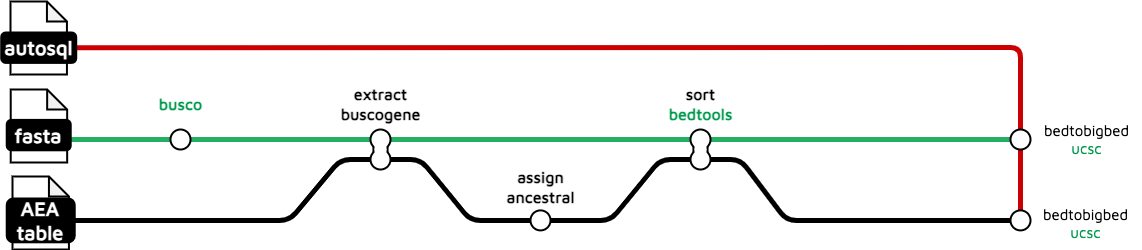
gene-alignment
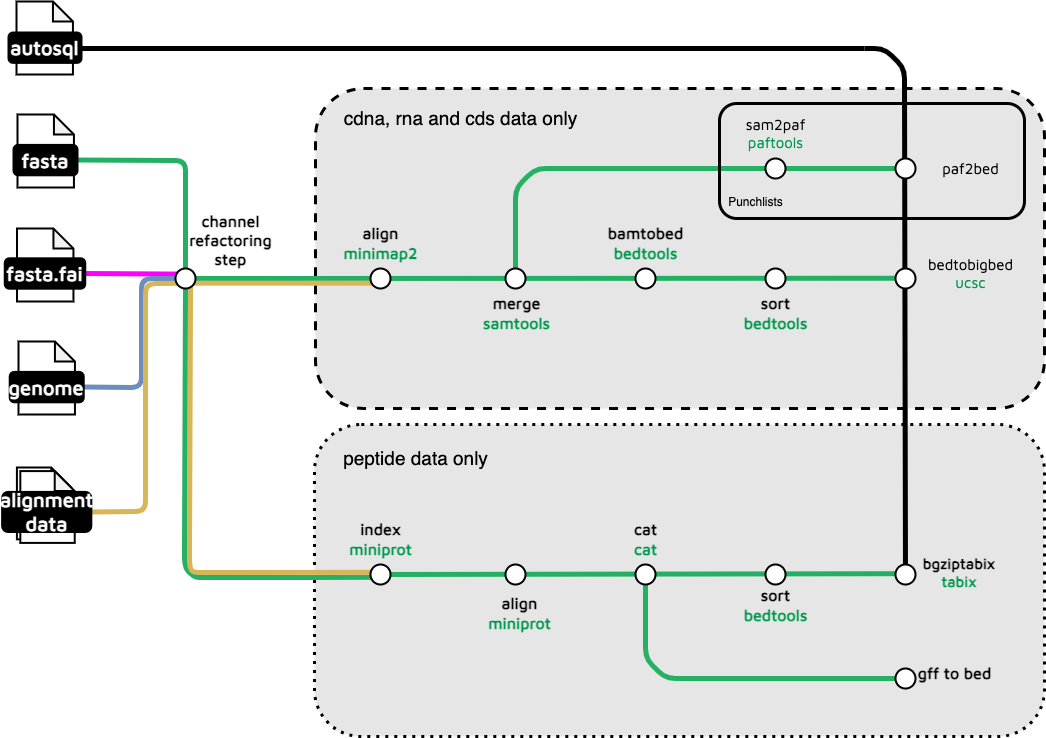
The gene alignment subworkflows load genesets (cdna, cds, rna, pep) data from a csv list of genomes, in the input .yaml, and aligns these to the reference genome. It contains two subworkflows, one of which handles peptide data and the other of which handles RNA, CDS and complementary DNA data. These produce files that can be displayed by JBrowse as tracks.
Output files
treeval_upload/*.gff.gz: Zipped .gff for each species with peptide data.*.gff.gz.tbi: TBI index file of each zipped .gff.*_cdna.bigBed: BigBed file for each species with complementary DNA data.*_cds.bigBed: BigBed file for each species with nuclear DNA data.*_rna.bigBed: BigBed file for each species with nRNA data.
treeval_upload/punchlists/*_pep_punchlist.bed: Punchlist for peptide track.*_cdna_punchlist.bed: Punchlist for cdna track.*_cds_punchlist.bed: Punchlist for cds track.*_rna_punchlist.bed: Punchlist for rna track.
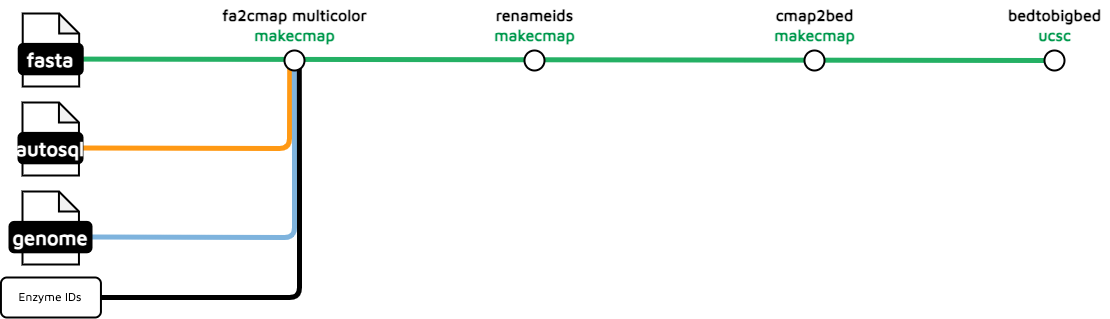
insilico-digest
The insilico-digest workflow is used to visualize the Bionano enzyme cutting sites for a genomic FASTA file. This procedure generates data tracks based on three digestion enzymes (by default): BSPQ1, BSSS1, and DLE1.
Output files
treeval_upload/{BSPQ1|BSSS1|DLE1}.bigBed: Bionano insilico digest cut sites track in the bigBed format for each of the set digestion enzymes.
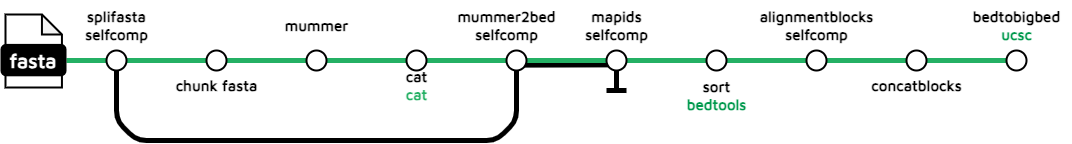
selfcomp
The selfcomp subworkflow is a comparative genomics analysis algorithm originally performed by the Ensembl projects database, and reverse engineered in Python3 by @yumisims. It involves comparing the genes and genomic sequences within a single species. The goal of the analysis is to identify haplotypic duplications in a particular genome assembly.
Output files
treeval_upload/*_selfcomp.bigBed: BigBed file containing selfcomp track data.
synteny
This subworkflow searches along a predetermined path for syntenic genome files based on clade and then aligns with MINIMAP2_ALIGN to the reference genome, emitting an aligned .paf file for each.
Output files
treeval_upload/*.paf: .paf file for each syntenic genomic aligned to reference.

kmer
This subworkflow performs a k-mer count using FASTK_FASTK then passes the results to MERQURYFK_MERQURYFK to plot a copy-number k-mer spectra.
Output files
hic_files/*.ref.spectra-cn.ln.png: .png file of copy number k-mer spectra.*.ktab*: K-mer table files.
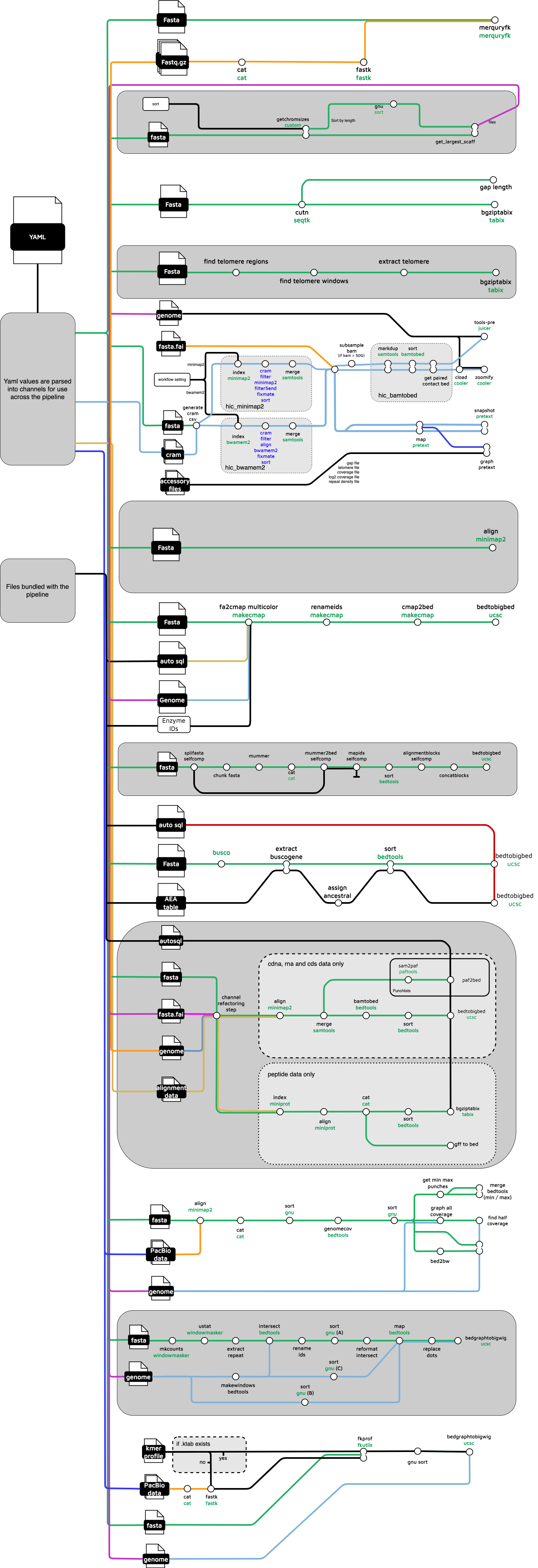
Full Workflow diagram
The full pipeline diagram is very large, with the pipeline consisting of over 100 processes.
pipeline-information
Nextflow provides excellent functionality for generating various reports relevant to the running and execution of the pipeline. This will allow you to troubleshoot errors with the running of the pipeline, and also provide you with other information such as launch commands, run times and resource usage.
Output files
treeval_info/TreeVal_Runs*.txt: A Report generated by TreeValProject.*txt: Reports generated by Nextflow:execution_report.html,execution_timeline.html,execution_trace.txtandpipeline_dag.dot/pipeline_dag.svg.- Reports generated by the pipeline:
pipeline_report.html,pipeline_report.txtandsoftware_versions.yml. Thepipeline_report*files will only be present if the--email/--email_on_failparameter's are used when running the pipeline. - Reformatted samplesheet files used as input to the pipeline:
samplesheet.valid.csv. - Parameters used by the pipeline run:
params.json.